Synthesis and Antiplasmodial Activity of Highly Active Reverse Analogues of the Antimalarial Drug Candidate Fosmidomycin
18-Aug-2010
ChemMedChem, 2010, DOI: 10.1002/cmdc.201000276, Volume 5, Issue 10, pages 1673–1676, published on 18.08.2010
ChemMedChem, online article
ChemMedChem, online article
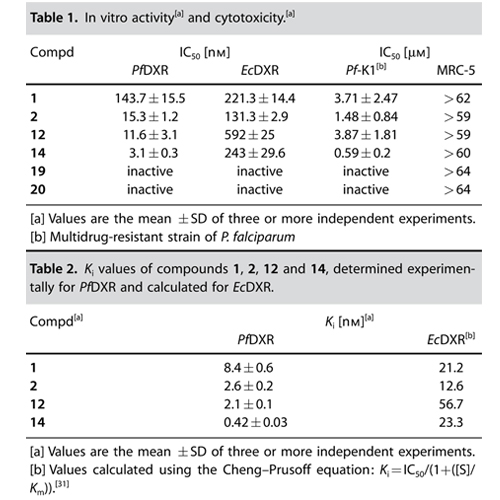
Although there are potent antimalarial drugs available, the spread of drug resistance has led to an increase in morbidity and mortality rates in malaria-endemic regions. To overcome these problems, new antimalarial drugs with novel modes of action have to be developed. The recently discovered nonmevalonate pathway of isoprenoid biosynthesis (MEP pathway) is an important metabolic pathway for the design of novel antimalarial drugs. Various pathogenic bacteria (e.g., Mycobacterium tuberculosis) and apicomplexan protozoa (e.g., Plasmodium falciparum) use the MEP pathway for the production of isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). IPP and DMAPP are important precursors for the biosynthesis of various essential isoprenoids. In contrast to the latter microorganisms, humans produce IPP and DMAPP exclusively via the mevalonate–acetate pathway. The second enzyme of the MEP pathway, 1-deoxy-d-xylulose 5-phosphate reductoisomerase (DXR, IspC) represents a validated target enzyme for the development of new antimalarial drugs. In 1998, the natural product fosmidomycin was identified as a potent inhibitor of P. falciparum DXR (PfDXR). The DXR catalyzes the rearrangement and reduction of 1-deoxy-d-xylulose 5-phosphate (DOXP) into 2-C-methyl-d-erythritol 4-phosphate (MEP). While the first step of the reaction requires a divalent cation (e.g., Mn2+ or Mg2+), the reduction step is NADPH-dependent. Clinical trials performed with fosmidomycin have already shown high efficiency in the treatment of acute, uncomplicated malaria tropica. However, the oral bioavailability of fosmidomycin is only moderate, with a resorption rate of about 10–30 %. Various research groups are involved in the design of novel DXR inhibitors. Molecular field analyses, as well as crystallographic studies, could already contribute in defining structural requirements for the development of potent DXR inhibitors. Consequently, different types of DXR inhibitors have been synthesized and biologically evaluated. Important structural elements for potent antimalarial activity are the hydroxamic acid functionality and the phosphonic acid group. The DXR–fosmidomycin complex crystal structure clearly revealed that the hydroxamic acid functionality chelates the divalent cation, while the phosphonic acid group occupies the phosphate binding site. Moreover, a defined distance between both functional groups is essential for potent antimalarial activity. Schlitzer and Klebe have shown that the two negative charges of the phosphonic acid group are necessary for high inhibitory activity. Furthermore, phosphonate prodrugs have been designed to improve the bioavailability of fosmidomycin (1) and FR900098 (2). A combinatorial approach for the synthesis of nonhydrolyzable phosphate mimics was reported by Link and co-workers, while Song recently described a coordination chemistry-based approach for the development of lipophilic DXR inhibitors that exhibit good to moderate activity against various pathogenic bacteria. To the best of our knowledge, a-aryl-substituted derivatives (4) of fosmidomycin are among the most active analogues known so far. Recently, Van Calenbergh reported that b-oxaisosters (3a,b) of fosmidomycin exhibit strong in vitro antimalarial activity. Interestingly, some of the new compounds contain an N-methyl-substituted hydroxamic acid group with a reverse orientation of the hydroxamate group (3b) compared to the lead compounds (1 and 2).