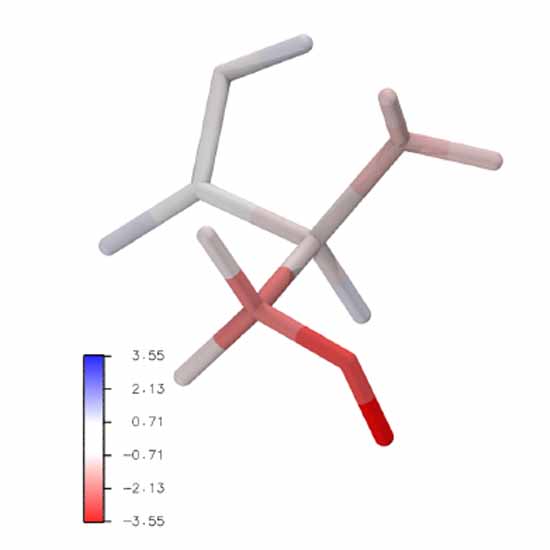

Calculating free energies from the vibrational density of states function: Validation and critical assessment
20-Mar-2019
J. Chem. Phys., 150, 194111 (2019); doi: 10.1063/1.5079643
J. Chem. Phys., online article
We explore and show the usefulness of the density of states function for computing vibrational free energies and free energy differences between small systems. Therefore, we compare this density of states integration method (DSI) to more established schemes such as Bennett’s Acceptance Ratio method (BAR), the Normal Mode Analysis (NMA), and the Quasiharmonic Analysis (QHA). The strengths and shortcomings of all methods are highlighted with three numerical examples. Furthermore, the free energy of the ionization of ammonia and the mutation from serine to cysteine are computed using extensive ab initio molecular dynamics simulations. We conclude that DSI improves upon the other frequency-based methods (NMA and QHA) regarding the treatment of anharmonicity and yielding results comparable to BAR in all cases without the need for alchemical transformations. Low-frequency modes lead to larger errors indicating that long simulation times might be required for larger systems. In addition, we introduce the use of DSI for the localization of the vibrational free energy to specific atoms or residues, leading to insights into the underlying process, a unique feature that is only offered by this method.