Time-Resolved Binding of Azithromycin to Escherichia coli Ribosomes
30-Jan-2009
J. Mol. Biol., 2009, 385(4), 1179-1192, doi:10.1016/j.jmb.2008.11.042 published on 30.01.2009
J. Mol. Biol., online article
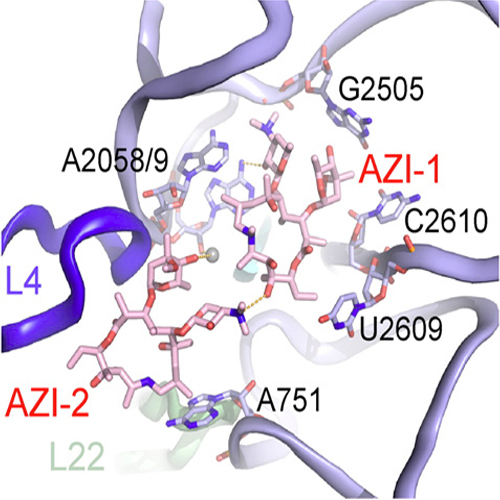
Azithromycin is a semisynthetic derivative of erythromycin that inhibits bacterial protein synthesis by binding within the peptide exit tunnel of the 50S ribosomal subunit. Nevertheless, there is still debate over what localization is primarily responsible for azithromycin binding and as to how many molecules of the drug actually bind per ribosome. In the present study, kinetic methods and footprinting analysis are coupled together to provide time-resolved details of the azithromycin binding process. It is shown that azithromycin binds to Escherichia coli ribosomes in a two-step process: The first-step involves recognition of azithromycin by the ribosomal machinery and places the drug in a low-affinity site located in the upper part of the exit tunnel. The second step corresponds to the slow formation of a final complex that is both much tighter and more potent in hindering the progression of the nascent peptide through the exit tunnel. Substitution of uracil by cytosine at nucleoside 2609 of 23S rRNA, a base implicated in the high-affinity site, facilitates the shift of azithromycin to this site. In contrast, mutation U754A hardly affects the binding process. Binding of azithromycin to both sites is hindered by high concentrations of Mg2+ ions. Unlike Mg2+ ions, polyamines do not significantly affect drug binding to the low-affinity site but attenuate the formation of the final complex. The low- and high-affinity sites of azithromycin binding are mutually exclusive, which means that one molecule of the drug binds per E. coli ribosome at a time. In contrast, kinetic and binding data indicate that in Deinococcus radiodurans, two molecules of azithromycin bind cooperatively to the ribosome. This finding confirms previous crystallographic results and supports the notion that species-specific structural differences may primarily account for the apparent discrepancies between the antibiotic binding modes obtained for different organisms.