CPD Damage Recognition by Transcribing RNA Polymerase II
09-Feb-2007
Science, 2007, 315, 859-62 (pdf) published on 09.02.2007
www.sciencemag.org, online article
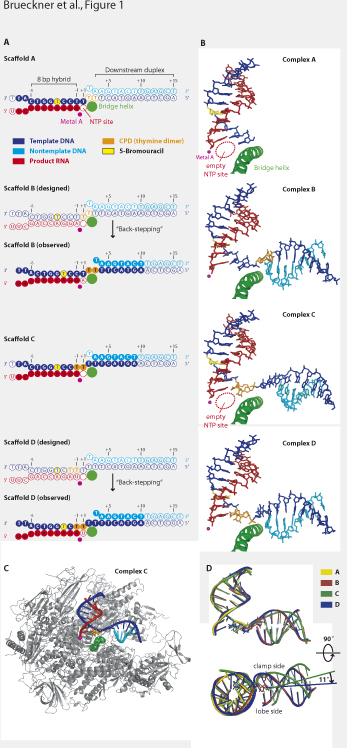
Cells use transcription-coupled repair (TCR) to efficiently eliminate DNA lesions such as UV-induced cyclobutane pyrimidine dimers (CPDs). Here we present the structure-based mechanism for the first step in eukaryotic TCR, CPD-induced stalling of RNA polymerase (Pol) II. A CPD in the transcribed strand slowly passes a translocation barrier, and enters the polymerase active site. The CPD 5’-thymine then directs uridine misincorporation into mRNA, which blocks translocation. Artificial replacement of the uridine by adenosine enables CPD bypass, thus Pol II stalling requires CPD-directed misincorporation. In the stalled complex, the lesion is inaccessible, and the polymerase conformation is unchanged. This is consistent with non-allosteric recruitment of repair factors and excision of a lesion-containing DNA fragment in the presence of Pol II. Ultraviolet light damages cellular DNA by inducing dimerization of adjacent pyrimidines in a DNA strand. The resulting cyclobutane pyrimidine dimer (CPD) lesions can block transcription and replication, and are a major cause of skin cancer (1). Cells eliminate CPDs by nucleotide excision repair (NER). A very efficient NER subpathway is transcription-coupled DNA repair (TCR), which specifically removes lesions from the DNA strand transcribed by Pol II (2). Pol II stalls when a CPD in the DNA template strand reaches the enzyme active site (3, 4). Pol II stalling apparently triggers TCR by recruitment of a transcription-repair coupling factor (Rad26 in yeast, CSB in humans), and factors required for subsequent steps of NER, including TFIIH, which comprises helicases that unwind DNA (5). Endonucleases then incise the DNA strand on either side of the lesion, resulting in a 24-34 nucleotide fragment (6-8). The obtained DNA gap is subsequently filled by DNA synthesis and ligation (9, 10). Fig. 1 Pol II elongation complex structures with thymine-thymine CPD lesions in the template. (A) Nucleic acid scaffolds A-D. The color code is used throughout. Filled circles denote nucleotides with interpretable electron density that were included in the structures in (B). Open circles denote nucleotides with non-interpretable or absent electron density. (B) Structure of nucleic acids in the Pol II elongation complexes A-D. The view is from the side (11). Figures prepared with PYMOL (DeLano Scientific). (C) Overview of complex C with a CPD lesion at the active site. The view is as in (B). Protein is in grey, the bridge helix in green. The CPD is shown as a stick model in orange. A large portion of the second largest Pol II subunit was omitted for clarity. (D) Superposition of nucleic acids in structures A-D. The protein molecules were superimposed and then omitted. The nucleic acids are depicted as ribbon models, the CPDs as stick models. Upper and lower views are related by a 90° rotation around a horizontal axis. To elucidate CPD recognition by transcribing Pol II, we carried out a structure-function analysis of elongation complexes containing in the template strand a thymine-thymine CPD. Elongation complexes were reconstituted from the 12-subunit Saccharomyces cerevisae Pol II and nucleic acid scaffolds as previously described (11), except that the mobile upstream DNA and the non-template strand in the transcription bubble were omitted (SOM text). A chemical analogue of a CPD lesion was incorporated at register +2/+3 of the template strand, directly downstream of position +1, which denotes the substrate addition site (scaffold A, Fig. 1A, SOM text) (12). The crystal structure of the resulting elongation complex A was determined (Fig. 1B) (12), and the register of the nucleic acids was unambiguously defined by bromine labeling (table S1, fig. S1C). The overall structure of complex A was nearly identical to the complete Pol II elongation complex (11) and very similar to elongation complex structures of the Pol II core enzyme (13, 14) (SOM text). The template strand enters the active site, and continues into an eight base pair hybrid duplex with RNA, which occupies the upstream positions –1 to –8 (Fig. 1B, SOM text). In contrast to the damage-free elongation complex (11), downstream DNA entering the cleft is mobile, indicating that the CPD at positions +2/+3 loosens the grip on downstream DNA (SOM text). To investigate Pol II stalling at the CPD lesion, we incubated complex A with nucleoside triphosphate (NTP) substrates, followed RNA extension by fluorescence-monitored capillary electrophoresis, and identified the RNA products by mass spectrometry (Figs. 2, 3) (12). After incubation with a physiological concentration of 1 mM NTPs for one hour, the RNA was extended by three nucleotides (Fig. 2A), but not any further. Thus the complex stalled after nucleotide incorporation opposite both CPD thymines. A time course showed that the first incorporation event was fast, consistent with a free substrate site in complex A (Figs. 1A, 1B, 2B, SOM text). The second and third incorporation events however were progressively slower, with rate constants of approx. 16 hr-1 and 2.4 hr-1, respectively (Fig. 2, A and B). Fig. 2 RNA extension assays (A) Electropherograms of time-dependent extension of RNA in complex A. A stoichiometric complex of complete Pol II and scaffold A (Fig. 1A) was incubated with 1 mM ATP, CTP, GTP and UTP. Reactions were stopped at the given time points. RNA products were subjected to fluorescence-monitored capillary electrophoresis, and identified by mass spectrometry. Signals for different RNAs are highlighted in different colors. (B) Quantification of time-dependent extension of RNA in complex A. Electropherogram signals in (A) were integrated and the relative amount of RNA product plotted against incubation time. (C) Specific uridine misincorporation opposite the CPD 5’-thymine. Stoichiometric complexes of complete Pol II and scaffold C were incubated with 1 mM of each NTP for 40 min. (D) Model for uridine misincorporation. In the upper panel, the structure of an undamaged Pol II elongation complex with a nonhydrolyzable NTP analogue (PDB 1Y77) was superposed on structure C. Depicted are the base pair at position +1 in 1Y77 (violet), and the CPD in structure C (orange). As modeled in the lower panel, the CPD 5’-thymine could form two hydrogen bonds with an incoming UTP. (E) Lesion bypass transcription. RNA in complex D (5’-AU-3’ opposite the CPD) was not extended after 20 min incubation with 1 mM of NTPs. Bypass was enabled under identical conditions by replacement of the RNA 3’-terminal uridine with adenosine (5’-AA-3’ opposite the CPD, scaffold DU→A, fig. S3). Structural considerations suggest that the second incorporation event is slower because translocation of the CPD from position +2/+3 to position +1/+2 is disfavored. Template bases in positions +1 and +2 are twisted against each other by 90° in the undamaged elongation complex (11), but twisting the CPD thymines is impossible since they are covalently linked. To test this, we included a CPD at positions +1/+2 of the scaffold, and solved the crystal structure of the resulting complex B (Fig. 1A, B). The CPD was observed at register +2/+3, indicating that it is not stably accommodated at positions +1/+2. Pol II had apparently “back-stepped” by one position, consistent with disfavored forward translocation (Fig. 3, SOM text). To efficiently overcome this translocation barrier, a concentration of 1 mM NTPs was required. Lower substrate concentrations limited RNA extension to one nucleotide after 5 min (fig. S2A). To investigate the very slow incorporation of the third nucleotide into complex A, we included a CPD at positions –1/+1, and solved the structure of the resulting complex C. The CPD was seen stably accommodated in the active site, and the NTP-binding site opposite the CPD 5’-thymine was free (Fig. 1A-C). We therefore used complex C to monitor incorporation of different NTPs. Only UTP led to nucleotide incorporation opposite the 5’-thymine (Fig. 2C), generally consistent with data for human Pol II (4). This misincorporation was very slow, with a rate constant of approx. 2.9 hr-1, comparable with the rate determined for the third nucleotide incorporation into complex A (fig. S2B, SOM text). Since translocation is not required for nucleotide incorporation in complex C, the rate-limiting step in reaching the stalled state is the slow uridine misincorporation, not CPD translocation from positions +1/+2 to –1/+1. Specific uridine misincorporation may be explained with the complex C structure. Whereas the CPD 3’-thymine occupies the same position as in the undamaged elongation complex (11), the CPD 5’-thymine is tilted by approx. 40°, and is shifted downwards by more than 2 Å into a wobble position, with the O4 atom at the location normally occupied by the N3 atom (Fig. 2D). Provided that binding of the incoming NTP (11, 14) is unaffected, the wobbled 5’-thymine could form two hydrogen bonds with UTP, whereas only one hydrogen bond would be possible with other NTPs (Fig. 2D). Attempts to visualize the CPD 5’-thymine-uridine mismatch crystallographically were unsuccessful (SOM text). These results suggested that Pol II stalls because translocation of the CPD 5’-thymine-uridine mismatch from position +1 to position –1 is strongly disfavored. This translocation event would move the damage-containing mismatch into the DNA-RNA hybrid, and the resulting distortion of the hybrid would destabilize the elongation complex (15) (SOM text). To test this model, we incorporated the CPD at positions –2/-1 of the scaffold, including a uridine residue opposite the 5’-thymine, and solved the structure of the resulting complex D (Fig. 1A, B). In this structure, Pol II had apparently back-stepped by one position, and the CPD was again located to positions –1/+1 in the active site, consistent with disfavored translocation (SOM text). Disfavored translocation of the CPD from position –1/+1 to –2/-1 may result from distortions due to the CPD and/or due to the mismatch. To distinguish these possibilities, we tested if Pol II extends the RNA in a variant of complex D with a matched CPD 5’-thymine-adenine base pair (Scaffold DU→A, fig. S3). Surprisingly, this RNA was extended to the run-off transcript (Fig. 2E, SOM text). Thus Pol II would bypass a CPD lesion if it could incorporate adenine opposite the CPD 5’-thymine. To test whether a T-U mismatch base pair alone is sufficient to stall Pol II, we used complex D, but without CPD, in RNA extension assays (Scaffold DΔCPD, Fig. S3). Only a small portion of the RNA was extended (fig. S2D). Taken together, Pol II stalling does not result from CPD-induced distortions per se, but from CPD-directed misincorporation. In contrast, DNA polymerases can correctly incorporate adenine opposite both CPD thymines, and, dependent on the type of polymerase, this can lead to stalling or lesion bypass (16, 17). Fig. 3 Mechanism of CPD recognition by transcribing Pol II. Schematic representation of RNA extension in complex A. The initial RNA (top) corresponds to the non-extended RNA of scaffold A. The translocation barrier and the translocation block are indicated with a dashed and a solid horizontal line, respectively. The artificial situation leading to lesion bypass (Fig. 2E) is depicted at the bottom. In all CPD-containing structures, the polymerase conformation is unchanged. This argues against allosteric models of TCR, which assume an incoming lesion causes a conformational change in Pol II that triggers recruitment of repair factors. In complexes B and D, downstream DNA is repositioned in the polymerase cleft (Fig. 1D). However, DNA repositioning cannot support an allosteric mechanism, since it occurs only in back-stepped complexes, which would not form when NTPs are present. A damage-stalled complex could alternatively be detected via exposure of the lesion by Pol II backtracking (18). The transcript cleavage factor TFIIS induces backtracking of a CPD-stalled complex (fig. S2E, SOM text), but TFIIS is not required for TCR in vivo (19). The lesion could also be exposed after polymerase bypass or dissociation from DNA. The latter mechanism underlies bacterial TCR, which involves the ATPase Mfd (20). However, the related eukaryotic ATPase CSB does neither trigger polymerase dissociation nor bypass (21). An alternative model for eukaryotic TCR that combines and extends previous models (7, 22, 23) can explain recognition of the stalled complex without allostery or exposure of the lesion (Fig. S4). Complexes that stall at an arrest site are rescued by TFIIS (24). Complexes that stall at a non-bulky lesion are rescued by CSB-induced lesion bypass (25). In both cases, transcription resumes. At a CPD lesion, however, CSB counteracts TFIIS-induced backtracking (26, 27), resulting in a stably stalled complex, and opening a time window for assembly of the repair machinery. TFIIH catalyzes extension of the transcription bubble (SOM text). This permits dual incision of the template strand on the Pol II surface (6, 7, 22). The lesion-containing DNA fragment and the RNA transcript are removed together with Pol II, although this requires more than dual incision (6-8, 28). The remaining gapped DNA is repaired. Pol II may be recycled, circumventing its ubiquitination and destruction (29). In conclusion, our data establish the molecular mechanism of CPD recognition by a cellular RNA polymerase, and provide a structural framework for further analysis of eukaryotic TCR.
References
- J. R. Mitchell, J. H. Hoeijmakers, L. J. Niedernhofer, Curr. Opin. Cell Biol. 15, 232 (2003).
- I. Mellon, G. Spivak, P. C. Hanawalt, Cell 51, 241-249.
- S. Tornaletti, B. A. Donahue, D. Reines, P. C. Hanawalt, J. Biol Chem. 272, 31719 (1997).
- J. S. Mei Kwei et al., Biochem. Biophys. Res. Commun. 320, 1133 (2004).
- T. T. Saxowsky, P. W. Doetsch, Chem. Rev. 106, 474 (2006).
- C. P. Selby, R. Drapkin, D. Reinberg, A. Sancar, Nucleic Acids Res. 25, 787 (1997).
- A. Tremeau-Bravard, T. Riedl, J. M. Egly, M. E. Dahmus, J. Biol. Chem. 279, 7751 (2004).
- D. Mu, A. Sancar, J. Biol. Chem. 272, 7570 (1997).
- A. Sancar, Annu. Rev. Biochem. 65, 43 (1996).
- S. Prakash, L. Prakash, Mutat. Res. 451, 13 (2000).
- H. Kettenberger, K.-J. Armache, P. Cramer, Mol. Cell 16, 955 (2004).
- Materials and methods are available as supporting material on Science Online.
- A. L. Gnatt, P. Cramer, J. Fu, D. A. Bushnell, R. D. Kornberg, Science 292, 1876 (2001).
- K. D. Westover, D. A. Bushnell, R. D. Kornberg, Cell 119, 481 (2004).
- M. L. Kireeva, N. Komissarova, D. S. Waugh, M. Kashlev, J. Biol. Chem. 275, 6530 (2000).
- H. Ling, F. Boudsocq, B. S. Plosky, R. Woodgate, W. Yang, Nature 424, 1083 (2003).
- Y. Li et al., Nat. Struct. Mol. Biol. 11, 784 (2004).
- B. A. Donahue, S. Yin, J. S. Taylor, D. Reines, P. C. Hanawalt, Proc. Natl. Acad. Sci. USA 91, 8502 (1994).
- R. A. Verhage, J. Heyn, P. van de Putte, J. Brouwer, Mol. Gen. Genet. 254, 284 (1997).
- A. M. Deaconescu et al., Cell 124, 507 (2006).
- C. P. Selby, A. Sancar, J. Biol. Chem. 272, 1885 (1997).
- A. H. Sarker et al., Mol. Cell 20, 187 (2005).
- J. Q. Svejstrup, J. Cell Sci. 116, 447 (2003).
- M. Wind, D. Reines, Bioessays 22, 327 (2000).
- S. K. Lee, S. L. Yu, L. Prakash, S. Prakash, Mol. Cell. Biol. 22, 4383 (2002).
- J. P. Laine, J. M. Egly, EMBO J. 25, 387 (2006).
- C. P. Selby, A. Sancar, Proc. Natl. Acad. Sci. USA 94, 11205 (1997).
- A. Aboussekhra et al., Cell 80, 859 (1995).
- E. C. Woudstra et al., Nature 415, 929 (2002).
- We thank H. Kettenberger, K. Armache, current members of the Cramer lab, A. Muschielok, J. Michaelis, and C. Schulze-Briese for help. This work was supported by the Deutsche Forschungsgemeinschaft, the Sonderforschungsbereich 646, the Volkswagen-Stiftung, and the Fonds der chemischen Industrie. Part of this work was performed at the Swiss Light Source at the Paul Scherrer Institute, Villigen, Switzerland. Structure coordinates and reflection files are deposited in the Protein Data Bank under accession numbers 2ja5, 2ja6, 2ja7, and 2ja8 for complexes A, B, C, and D, respectively.
Munich Center for Integrated Protein Science CIPSMDepartment of Chemistry and Biochemistry and Gene Center MunichLudwig-Maximilians-Universität MünchenFeodor-Lynen-Str. 25, 81377 Munich, Germany
*Correspondence: thomas.carell@cup.uni-muenchen.de, cramer@lmb.uni-muenchen.de