ProteomeTools: Systematic characterization of 21 post-translational protein modifications by LC-MS/MS using synthetic peptides
29-May-2018
Molecular Cellular Proteomics, doi: 10.1074/mcp.TIR118.000783 mcp.TIR118.000783
Molecular Cellular Proteomics, online ablage
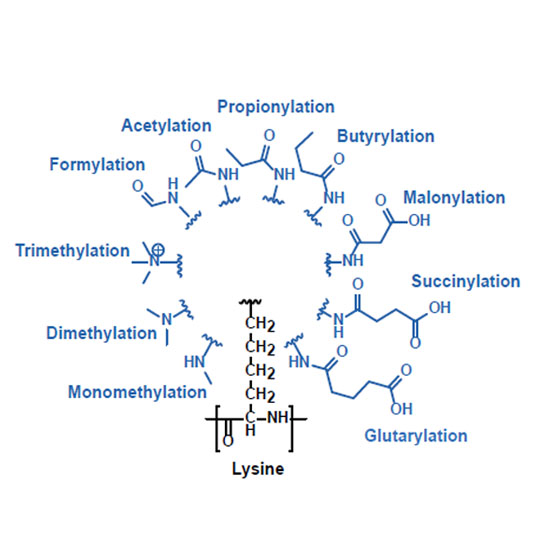
The analysis of the post-translational modification (PTM) state of proteins using mass spectrometry-based bottom-up proteomic workflows has evolved into a powerful tool for the study of cellular regulatory events that are not directly encoded at the genome level. Besides frequently detected modifications such as phosphorylation, acetylation and ubiquitination, many low abundant or less frequently detected PTMs are known or postulated to serve important regulatory functions. To more broadly understand the LC-MS/MS characteristics of PTMs, we synthesized and analyzed ~5,000 peptides representing 21 different naturally occurring modifications of lysine, arginine, proline and tyrosine side chains and their unmodified counterparts. The analysis identified changes in retention times, shifts of precursor charge states and differences in search engine scores between modifications. PTM-dependent changes in the fragmentation behavior were evaluated using eleven different fragmentation modes or collision energies. We also systematically investigated the formation of diagnostic ions or neutral losses for all PTMs, confirming 10 known and identifying 5 novel diagnostic ions for lysine modifications. To demonstrate the value of including diagnostic ions in database searching, we reprocessed a public data set of lysine crotonylation and showed that considering the diagnostic ions increases confidence in the identification of the modified peptides. To our knowledge, this constitutes the first broad and systematic analysis of the LC-MS/MS properties of common and rare PTMs using synthetic peptides, leading to direct applicable utility for bottom-up proteomic experiments.