Synthetic and structural studies on syringolin A and B reveal critical determinants of selectivity and potency of proteasome inhibition
21-Apr-2009
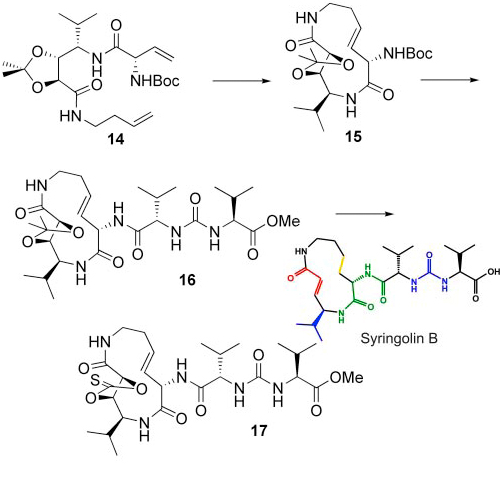
Syrbactins, a family of natural products belonging either to the syringolin or glidobactin class, are highly potent proteasome inhibitors. Although sharing similar structural features, they differ in their macrocyclic lactam core structure and exocyclic side chain. These structural variations critically influence inhibitory potency and proteasome subsite selectivity. Here, we describe the total synthesis of syringolin A and B, which together with enzyme kinetic and structural studies, allowed us to elucidate the structural determinants underlying the proteasomal subsite selectivity and binding affinity of syrbactins. These findings were used successfully in the rational design and synthesis of a syringolin A-based lipophilic derivative, which proved to be the most potent syrbactin-based proteasome inhibitor described so far. With a Ki′ of 8.65 ± 1.13 nM for the chymotryptic activity, this syringolin A derivative displays a 100-fold higher potency than the parent compound syringolin A. In light of the medicinal relevance of proteasome inhibitors as anticancer compounds, the present findings may assist in the rational design and development of syrbactin-based chemotherapeutics.