Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome
15-Apr-2014
PNAS, 2014, doi: 10.1073/pnas.1403409111, vol. 111 no. 15, 5544–5549, published on 15.04.2014
PNAS, online article
PNAS, online article
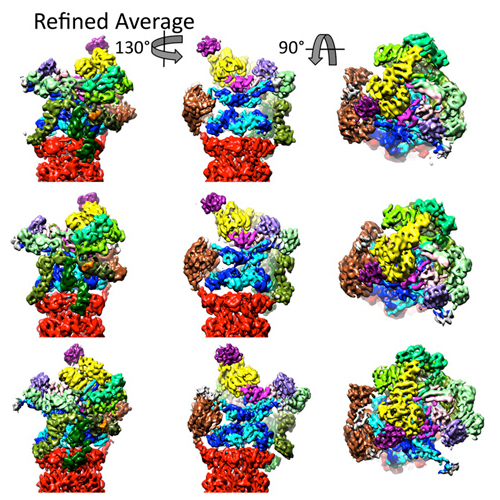
The 26S proteasome is a 2.5 MDa molecular machine that executes the degradation of substrates of the ubiquitin–proteasome pathway. The molecular architecture of the 26S proteasome was recently established by cryo-EM approaches. For a detailed understanding of the sequence of events from the initial binding of polyubiquitylated substrates to the translocation into the proteolytic core complex, it is necessary to move beyond static structures and characterize the conformational landscape of the 26S proteasome. To this end we have subjected a large cryo-EM dataset acquired in the presence of ATP and ATP-γS to a deep classification procedure, which deconvolutes coexisting conformational states. Highly variable regions, such as the density assigned to the largest subunit, Rpn1, are now well resolved and rendered interpretable. Our analysis reveals the existence of three major conformations: in addition to the previously described ATP-hydrolyzing (ATPh) and ATP-γS conformations, an intermediate state has been found. Its AAA-ATPase module adopts essentially the same topology that is observed in the ATPh conformation, whereas the lid is more similar to the ATP-γS bound state. Based on the conformational ensemble of the 26S proteasome in solution, we propose a mechanistic model for substrate recognition, commitment, deubiquitylation, and translocation into the core particle.