Phosphorylation of histone H3T6 by PKCbI controls demethylation at histone H3K4
14-Mar-2010
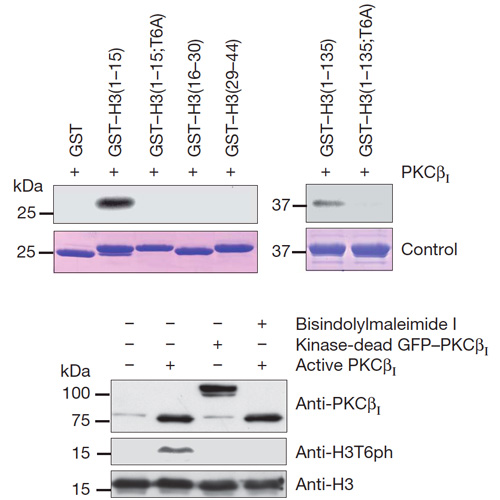
Demethylation at distinct lysine residues in histone H3 by lysinespecific demethylase 1 (LSD1) causes either gene repression or activation1,2. As a component of co-repressor complexes, LSD1 contributes to target gene repression by removing mono- and dimethyl marks from lysine 4 of histone H3 (H3K4)1,3. In contrast, during androgen receptor (AR)-activated gene expression, LSD1 removes mono- and dimethyl marks from lysine 9 of histone H3 (H3K9)2. Yet, the mechanisms that control this dual specificity of demethylation are unknown. Here we show that phosphorylation of histone H3 at threonine 6 (H3T6) by protein kinase C beta I (PKCbI, also known as PRKCb) is the key event that prevents LSD1 from demethylating H3K4 during AR-dependent gene activation. In vitro, histone H3 peptides methylated at lysine 4 and phosphorylated at threonine 6 are no longer LSD1 substrates. In vivo, PKCbI co-localizes with AR and LSD1 on target gene promoters and phosphorylates H3T6 after androgen-induced gene expression.RNA interference (RNAi)-mediated knockdown of PKCbIabrogates H3T6 phosphorylation, enhances demethylation at H3K4, and inhibits AR-dependent transcription. Activation of PKCbI requires androgen-dependent recruitment of the gatekeeper kinase protein kinase C (PKC)-related kinase 1 (PRK1)4. Notably, increased levels of PKCbI and phosphorylated H3T6 (H3T6ph) positively correlate with high Gleason scores of prostate carcinomas, and inhibition of PKCbI blocks AR-induced tumour cell proliferation in vitro and cancer progression of tumour xenografts in vivo. Together, our data establish that androgen-dependent kinase signalling leads to the writing of the new chromatin mark H3T6ph, which in consequence prevents removal of active methyl marks from H3K4 during AR-stimulated gene expression.